

シニア・リサーチ・サイエンティストである Sanchit Misra は、インテルラボの計算生物学/HPC 研究の取り組みを率いています。この記事は、同じくリサーチ・サイエンティストで、インテルラボで計算生物学/HPC 研究を行っている Narendra Chaudhary との共著です。
ハイライト
- シングルセル RNA-Seq (scRNA-Seq) 解析は、単一細胞の遺伝子発現を解読することで、細胞レベルでの遺伝子の洞察を可能にする、単一細胞解析の先進的なアプローチです。
- インテルラボは、scRNA-Seq 解析のための Scanpy ベースのパイプラインを CPU ベースラインよりも約 40 倍高速化し、GCP 上のシングル CPU インスタンスで 130 万個のマウス細胞の解析をわずか 7 分半で達成しました。これは、A100 GPU を 1 台使用した場合の約 5 倍の性能で、並列アルゴリズム、いくつかのアーキテクチャー固有の最適化、インテル® oneDAL や Katana Graph などの基礎となるライブラリーの強化により、それぞれのチームとの密接な協力のもとで達成されたものです。
計測の分解能の飛躍的な向上は、常に分野に革命をもたらしてきました。例えば、顕微鏡や望遠鏡の発明が科学に与えたインパクトはとても大きいものでした。シングルセル解析は、生物学において同様の革命が展開されている重要な一例となっています。人間の身体は 40 兆個近い細胞からできています。以前はこれらの細胞はまとめて、(ときには数百万個の細胞を一度に) 調べてきたため、細胞間の違いを捉えることができませんでした。先進的なアプローチである単一細胞解析は、細胞の個性を研究する分野です。新しい細胞タイプの特定、これらの細胞を互いに異なるものにするメカニズムの解明、特定の病気や薬剤に対する細胞の反応の実証など、細胞分化の謎を解き明かし始めています。この、比較的新しい分野は、がんから Covid-19 関連の研究に至るまで、生物学的発見への計り知れない可能性をすでに示しています。
データ計測技術の進歩により、シングルセルのデータ量は急速に増加しています。個々のデータセットのサイズも同様の速度で増えてきています。このようなデータ解析には、通常、データ・サイエンス・パイプラインを実行する必要があります。パイプラインのステップは、パラメーターを変更しながら繰り返されることが多いため、ほぼリアルタイムで実行できる対話型のパイプラインが有効とされています。
シングル CPU で 130 万個のマウス細胞の ScRNA-seq 解析をわずか 7 分半で達成
細胞分化のさまざまな側面を研究するために、多くの種類のシングルセル解析があります。シングルセル RNA-seq (scRNA-seq) 解析は、細胞間の遺伝子発現プロファイルの違いを研究します。しかしこれは、個々の細胞の遺伝子発現を測定することができる高度な技術であるシングルセル RNA シーケンスに依存します。
scRNA-seq 解析の典型的なワークフローは、各細胞の遺伝子の発現量からなる行列から始まります。データ前処理のステップでは、ノイズを除去し、データを正規化して、データセットの個々の細胞におけるすべてのヒト遺伝子の活性を得ることができます。このステップでは、データ収集から生じるアーティファクトを修正するために、機械学習がしばしば利用されます。その後、次元削減が行われ、類似した遺伝子活性を持つ細胞をグループ化するためのクラスタリングが行われ、クラスターが可視化されます。Scanpy は、80 万回以上ダウンロードされている、この解析のために最も広く使われているツールキットの 1 つです。
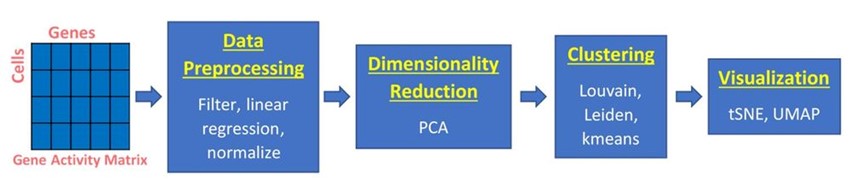
図 1: 単一細胞 RNA シーケンスデータの解析手順を示すパイプライン (遺伝子活性行列から始まり、異なる細胞クラスターの可視化まで)
130 万個のマウス脳細胞からなるデータセットでは、既製の (ベースラインの) Scanpy 実装を使用した GCP のシングル CPU インスタンス (n1-highmem-64) において、図 1 に示したパイプラインは通常 5 時間近くかかると思われます。しかし、同じパイプラインで、Nvidia は Nvidia RAPIDS を使用して、単一の A100 GPU 上でエンドツーエンドの実行時間は 686 秒であったと報告しています。
インテルラボでは、インテル® oneDAL チームと Katana Graph が協力して、より優れた並列アルゴリズムを使用してパイプラインを高速化し、基礎となるアーキテクチャーに合わせてパフォーマンスをチューニングしました。これはまだ作業中ですが、以下の表とグラフは、現在のパフォーマンスとクラウド利用コストを示しています。これらの結果は、先日開催された Intel Investor Day 2022 で発表されました。GCP 上の同じシングル CPU インスタンス (n1-highmem-64) 上で、パイプライン全体をわずか 626 秒で終了できるようになったのです。このパフォーマンスは、第 3 世代インテル® Xeon® スケーラブル・プロセッサー (開発コード名: Ice Lake) で実行する新しい n2 インスタンス・タイプでさらに向上しています。また、パイプラインのメモリー要件も変更し、ハイメモリー n2-highmem-64 インスタンスの代わりにローメモリー n2-highcpu-64 インスタンスを使用できるようにしました。GCP 上の n2-highcpu-64 のシングル・インスタンスでは、パイプライン全体がわずか 459 秒 (7.65 分) で終了します。これは、最初に設定した 5 時間の CPU ベースラインと比較して、約 40 倍の速さです。また、Nvidia A100 のパフォーマンスと比べても 1.5 倍近い速さです。
高速化とメモリー要件の変更により、クラウドコストを大幅に削減することができました。表にあるように、GCP 上の n2-highcpu-64 インスタンスのコストはわずか $0.29 です。これは、ベースライン Scanpy を実行する n1-highmem-64 の約 66 分の 1、Nvidia A100 GPU の半分以下です。
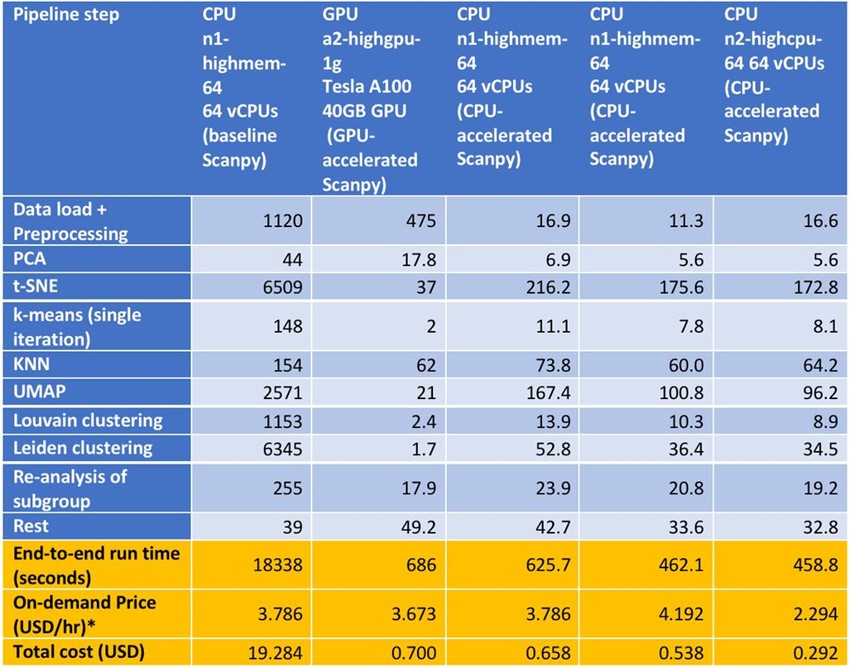
表 1: さまざまな GCP インスタンスにおける 130 万個のマウス脳細胞の scRNA-seq 解析の実行時間とクラウドコストを示しています。最初の 2 列は、シングル CPU インスタンス (n1-highmem-64) 上のベースライン Scanpy とシングル GPU インスタンス (a2-highgpu-1g) 上の GPU アクセラレーション Scanpy の実行時間とクラウドコストを表しています。最後の 3 列は、2 世代の CPU インスタンス・タイプ (n1-highmem-64、n1-highmem-64、n2-highcpu-64) のシングル・インスタンス上での CPU アクセラレーションによる Scanpy の実行時間およびクラウドコストの測定値**のレポートです。
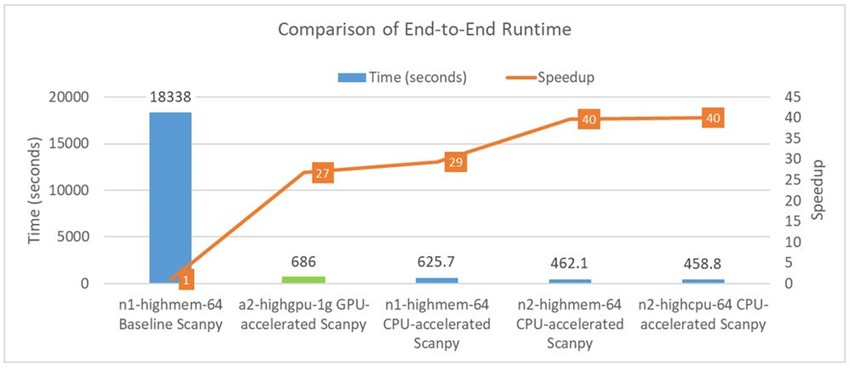
図 2: さまざまな GCP インスタンスにおける 130 万個のマウス脳細胞の scRNA-seq 解析の実行時間と高速化を表しています。グラフは、(1) シングル CPU インスタンス (n1-highmem-64) 上でのベースライン Scanpy とシングル GPU インスタンス (a2-highgpu-1g) 上での GPU アクセラレーション Scanpy の公開実行時間、(2) 2 世代の CPU インスタンス・タイプ (n1-highmem-64、n2-highmem-64、n2-highcpu-64) 上での CPU アクセラレーション Scanpy の測定実行時間** を使用しています。また、折れ線グラフは、n1-highmem-64 インスタンス上で動作するベースライン Scanpy に対する高速化を示しています。
** 2022年5月25日時点のインテルによるテスト
データ・サイエンス・パイプラインをどのように高速化したか
このパイプラインのパフォーマンスを向上させるために行った処理を以下に詳述します。
- データ前処理の効率を上げるため、ウォーム・ファイル・キャッシュを使用し、JIT (ジャストインタイム) コンパイラーである Numba を用いてマルチスレッド化しました。これにより、ベースラインの前処理パフォーマンスを 70 倍以上向上させることができました。
- また、K-means クラスタリング、KNN (K Nearest Neighbor)、PCA (Principal Component Analysis) が効率的に実装された scikit-learn 向けインテル® エクステンションを使用しました。
- Scanpy はもともと scikit-learn の tSNE (t-distributed Stochastic Neighbor Embedding) 実装を使用していましたが、インテル® Xeon® プロセッサーでは非効率でした。そこでインテル® Xeon® プロセッサーで効率の良い実装を構築することで、tSNE の 40 倍近い高速化を達成しました。
- Barnes-Hut アルゴリズムの共有メモリー並列実装
- Morton コードを用いた 4 分木の構築、ソート、要約ステップの並列化
- 以下のように UMAP (Uniform Manifold Approximation and Projection) を最適化しました。
- Python ソースコードから C++ への変換
- インテル® AVX-512/インテル® AVX 2 ベースの疑似乱数生成器の効率的な実装の作成
- 固有値計算ステップにインテル® oneAPI マス・カーネル・ライブラリー (インテル® oneMKL) を使用
- Katana Graph チームは、このコラボレーションの一環として、Louvain と Leiden のアルゴリズムを効率的に実装し、パイプラインに統合しました。
これらの開発により、大規模なデータセットの解析にかかる時間が大幅に短縮され、CPU で 40 倍、Nvidia A100 GPU で 1.5 倍の速さで作業を完了できるようになりました。
結論
シングルセル解析は、腫瘍学、微生物学、神経学、生殖学、免疫学、消化器系、泌尿器系など、多くの分野で応用されています。うまくいけば、作業時間の短縮によってさまざまな細胞をより深く理解できるようになり、大きな利益をもたらす医学の進歩への道を開くことができます。インテルは、scRNA-seq 解析パイプラインをさらに洗練させることに取り組んでいます。具体的には、tSNE、UMAP、Leiden のさらなる改良に注力しています。
構築環境の詳細
GCP n1-highmem-64: 1-instance GCP n1-highmem-64: 64 vCPUs (Skylake), 416 GB total memory, bios: Google, ucode: 0x1, Ubuntu 20.04, 5.13.0-1024-gcp
GCP n2-highmem-64: 1-instance GCP n2-highmem-64: 64 vCPUs (Ice Lake), 512 GB total memory, bios: Google, ucode: 0x1, Ubuntu 20.04, 5.13.0-1024-gcp
GCP n2-highcpu-64: 1-instance GCP n2-highcpu-64: 64 vCPUs (Ice Lake), 64 GB total memory, bios: Google, ucode: 0x1, Ubuntu 20.04, 5.13.0-1024-gcp
参照記事:
Intel Labs Accelerates Single-cell RNA-Seq Analysis